News | ASCO
25.07.2022
Ewing-Tumor: Therapie orientiert sich am Ewing-Protokoll
Inhaltsverzeichnis
[Verbergen]
Anzeige:

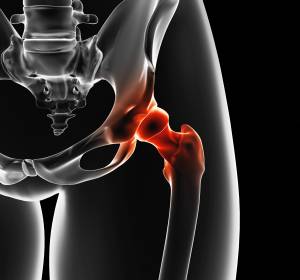
Zu den Ewing-Tumoren zählen das Ewing-Sarkom, der primitive neuroektodermale Tumor (PNET), der Askin-Tumor sowie das extra-skelettale Ewing-Sarkom.
Das Ewing-Sarkom ist bei Kindern nach dem Osteosarkom der zweithäufigste Knochentumor und endet trotz moderner Therapien immer noch oft tödlich. Besonders, wenn bei Diagnose schon Metastasen gebildet wurden, ist die Prognose für die jungen Patienten schlecht.
Als erstes Symptom sind bei Extremitätenbefall häufig lokale Schwellung oder Schmerzen zu verzeichnen. Tumoren im Becken und Thorax wachsen lange unerkannt.
Die Diagnose von Ewing-Tumoren kann in den meisten Fällen bereits mittels Röntgenbefund gestellt werden. Anhand von Magnetresonanztomographie (MRT) und Computertomographie (CT) lassen sich die Lage und das Ausmaß des Tumors einschätzen. Bei Patienten in einem Alter zwischen 48 Monaten und 50 Jahren orientiert sich das Vorgehen bei der Therapie am Ewing-2008-Protokoll. Dieses strebt an, die Behandlungsergebnisse für Patienten zu optimieren. Weitere Informationen hierzu gibt es unter: www.kinderkrebsinfo.de. Je nach Risikoabschätzung wird der Betroffene mehr oder weniger intensiv behandelt. Für Patienten jenseits des 50. Lebensjahres existiert kein allgemein anerkannter Standard. Diese Patienten können z.B. mit Adriamycin und Ifosfamid behandelt werden.
Das Ewing-Sarkom ist bei Kindern nach dem Osteosarkom der zweithäufigste Knochentumor und endet trotz moderner Therapien immer noch oft tödlich. Besonders, wenn bei Diagnose schon Metastasen gebildet wurden, ist die Prognose für die jungen Patienten schlecht.
Als erstes Symptom sind bei Extremitätenbefall häufig lokale Schwellung oder Schmerzen zu verzeichnen. Tumoren im Becken und Thorax wachsen lange unerkannt.
Die Diagnose von Ewing-Tumoren kann in den meisten Fällen bereits mittels Röntgenbefund gestellt werden. Anhand von Magnetresonanztomographie (MRT) und Computertomographie (CT) lassen sich die Lage und das Ausmaß des Tumors einschätzen. Bei Patienten in einem Alter zwischen 48 Monaten und 50 Jahren orientiert sich das Vorgehen bei der Therapie am Ewing-2008-Protokoll. Dieses strebt an, die Behandlungsergebnisse für Patienten zu optimieren. Weitere Informationen hierzu gibt es unter: www.kinderkrebsinfo.de. Je nach Risikoabschätzung wird der Betroffene mehr oder weniger intensiv behandelt. Für Patienten jenseits des 50. Lebensjahres existiert kein allgemein anerkannter Standard. Diese Patienten können z.B. mit Adriamycin und Ifosfamid behandelt werden.
Rezidiv oder Krankheitsprogress
Im Falle eines Rezidivs oder einer Krankheitsprogression gibt es keinen einheitlichen Standard. Es kommen lokaltherapeutische Maßnahmen zum Einsatz oder eine Rezidiv-Chemotherapie. Seit 2009 erfolgt in Deutschland die systemische Erfassung von Sarkom-Rezidiven im Rahmen des Netzwerkprojekts TranSaRNet. Die Heilungschance bei einem vorliegenden Rezidiv ist gering mit einem 2-Jahres-Überleben von nur etwa 20%.Aktuelle Grundlagenforschung
Neuere Forschungen zu der Tumorentstehung beim Ewing-Sarkom konnten eine einzige dominante Mutation identifizieren, die die Hauptursache sein könnte. Eine angeborene Genvariabilität könnte der Grund für die unterschiedlichen Verläufe der Erkrankung darstellen.Redaktion von journalonko.de (SK)

Beiträge zum Thema: Ewing-Sarkom

News
06.06.2022

News
12.05.2022

Anzeige:

News
22.03.2022

Fortbildung
25.01.2022

News
09.09.2021
Sarkome
25.06.2021

News | ASCO
07.06.2021

News
07.06.2021

Sarkome
16.09.2020
Sarkome
16.09.2020
News
12.09.2019
News
10.07.2019
News
05.06.2019

Sarkome
24.05.2019
Sarkome
24.05.2019
Sarkome
24.05.2019

News
21.11.2018
News
21.06.2018

Fortbildung
15.02.2018